Annexe 1 des BPF – Critères de sélection des vêtements de protection pour salle blanche
Par Steve Marnach de DuPont.
Pour la première fois depuis 2008, l’annexe 1 des directives européennes sur les bonnes pratiques de fabrication (BPF) pour la fabrication de médicaments stériles a fait l’objet d’une révision en profondeur. La nouvelle annexe 1 des BPF a été publiée le 25 août 2022. Elle représente bien plus qu’une simple mise à jour. La directive actuelle a été complètement réécrite et non seulement la longueur de l’annexe 1 a été augmentée de 16 à 58 pages, mais toute l’approche qui y est décrite a été modifiée. Cela aura des répercussions tant sur les technologies que sur les procédures utilisées dans la fabrication pharmaceutique.
L’extrait suivant de la section 2.2 à la page 4 résume la nouvelle approche :
“Les processus, les équipements, les installations et les activités de fabrication doivent être gérés conformément aux principes QRM (Quality Risk Management, gestion du risque qualité) qui fournissent un moyen proactif d’identification, d’évaluation scientifique et de contrôle des risques potentiels pour la qualité. (…) La surveillance ou les tests seuls ne garantissent pas la stérilité.”
Il est prévu que toutes les activités au sein de la fabrication pharmaceutique seront régies de manière holistique par les principes QRM et documentées dans la stratégie de contrôle de la contamination (CCS). Nous nous attendons à ce que la CCS soit un document évolutif, basé sur une approche scientifique axée sur les données, qui devrait être continuellement mis à jour et amélioré afin de contrôler les risques potentiels pour la qualité. La nouvelle annexe 1 des BPF appelle à une approche proactive et simplement réagir et corriger la contamination détectée ne suffira plus. Il est attendu des fabricants qu’ils comprennent parfaitement leurs processus et procédures, de sorte qu’ils puissent identifier les risques potentiels pour la qualité, mettre en place tous les moyens techniques et procéduraux nécessaires pour maîtriser ces risques et atteindre des améliorations continues. Puisque les systèmes vestimentaires de salle blanche sont un élément essentiel de la fabrication stérile et aseptique, ils doivent également être gérés selon les principes QRM.
Principes QRM pour les vêtements de salle blanche
La QRM commence par une analyse complète basée sur des données et une compréhension de tous les risques pour la qualité liés aux opérateurs de salle blanche portant des vêtements de salle blanche. Un tel examen permettra à l’analyste de concevoir des procédures de certification, de qualification, de validation et de surveillance intégrant la qualité, faisant partie d’une CCS holistique. Une analyse des risques est nécessaire pour comprendre les risques de contamination provenant des opérateurs portant des vêtements de salle blanche. Les opérateurs représentent la plus grande source de contamination à l’intérieur des salles blanches, entraînant 75 % de tous les contaminants.1,2 Cette contamination provient à la fois des opérateurs eux-mêmes et de leurs vêtements de salle blanche.
La contamination “humaine” provenant des opérateurs est due à la fois à la biologie (une personne moyenne libère 40 000 particules par minute et 10 % de celles-ci sont porteuses de microorganismes) et comportementale. Bien qu’il soit possible d’atténuer ce dernier grâce à une sélection minutieuse des opérateurs, une formation, des mouvements lents et une hygiène irréprochable, le fait est (comme de nombreuses études l’ont prouvé) que les opérateurs rejetteront toujours des particules. La seule mesure pour empêcher les particules générées par les opérateurs de contaminer la salle blanche est le port de vêtements de salle blanche, qui sont la seule barrière entre l’opérateur et l’environnement de production. L’annexe 1 des BPF 2022 l’indique clairement à la section 7.13 i.
"Les vêtements de protection doivent minimiser la perte de fibres ou de particules et retenir les particules rejetées par le corps."
Comme indiqué ci-dessus, les vêtements de salle blanche eux-mêmes peuvent être une source de contamination et ce risque doit également être évalué. Par exemple, le matériau utilisé pour la confection des vêtements (non-tissé pour les vêtements à usage unique ou tissé pour les articles réutilisables) peut rejeter un nombre plus ou moins important de particules selon la nature des fibres ou filaments utilisés, leur résistance à l’abrasion ou leur construction. Les garnitures (fermetures éclair, boutons, élastiques ou fils à coudre) peuvent également être une source de contamination, tandis que la conception du vêtement peut également jouer un rôle, et ceux-ci doivent également être évalués. Un détail souvent négligé est l’emballage dans lequel les vêtements de salle blanche arrivent, qui pourrait également être une source de contamination (c’est-à-dire les sacs en papier par rapport aux sacs en plastique).
Principales étapes de validation
Une fois les risques évalués, les causes de ces risques devraient être supprimées ou remplacées par des moyens techniques ou organisationnels dans la mesure du possible. Les risques résiduels doivent être atténués autant que possible en utilisant un système de vêtements de salle blanche validé. Les directives générales de l’UE sur la validation (annexe 1519 des BPF) fournissent un cadre général qui peut être appliqué à la qualification des systèmes de vêtements de salle blanche. Cette approche de validation comprend cinq étapes : la définition d’une spécification des exigences de l’utilisateur (URS), une qualification de conception (DQ), une qualification d’installation (IQ), une qualification opérationnelle (OQ) et une qualification de performance (PQ). Si la DQ et l’IQ ont le plus d’impact sur la qualité atteinte, les autres étapes ne doivent pas être négligées et il est important de procéder étape par étape.3
Spécification des exigences de l’utilisateur (URS)
Bien qu’ils ne fassent pas officiellement partie du processus de validation, il est important de définir dès le départ les exigences relatives à un système de vêtements de salle blanche à partir de l’utilisateur et de l’environnement dans lequel il travaille. L’URS définira les exigences critiques par rapport auxquelles le système vestimentaire doit être évalué de manière à ce qu’il soit conforme à l’évaluation des risques. Par exemple, un opérateur formé peut devoir être capable de travailler pendant au moins 3 heures dans le même ensemble de vêtements de salle blanche sans provoquer des niveaux inacceptables de contamination des vêtements et de l’environnement de travail aseptique (selon les BPF actuelles [cGMP]). Le système d’emballage du vêtement doit être adapté à l’aménagement de la salle blanche et à son système de passage de matériel, ou peut être adapté à une désinfection manuelle par pulvérisation. L’opérateur peut également parfois avoir besoin d’une protection chimique ou biologique contre les substances qu’il manipule dans la salle blanche.
Qualification de conception (DQ)
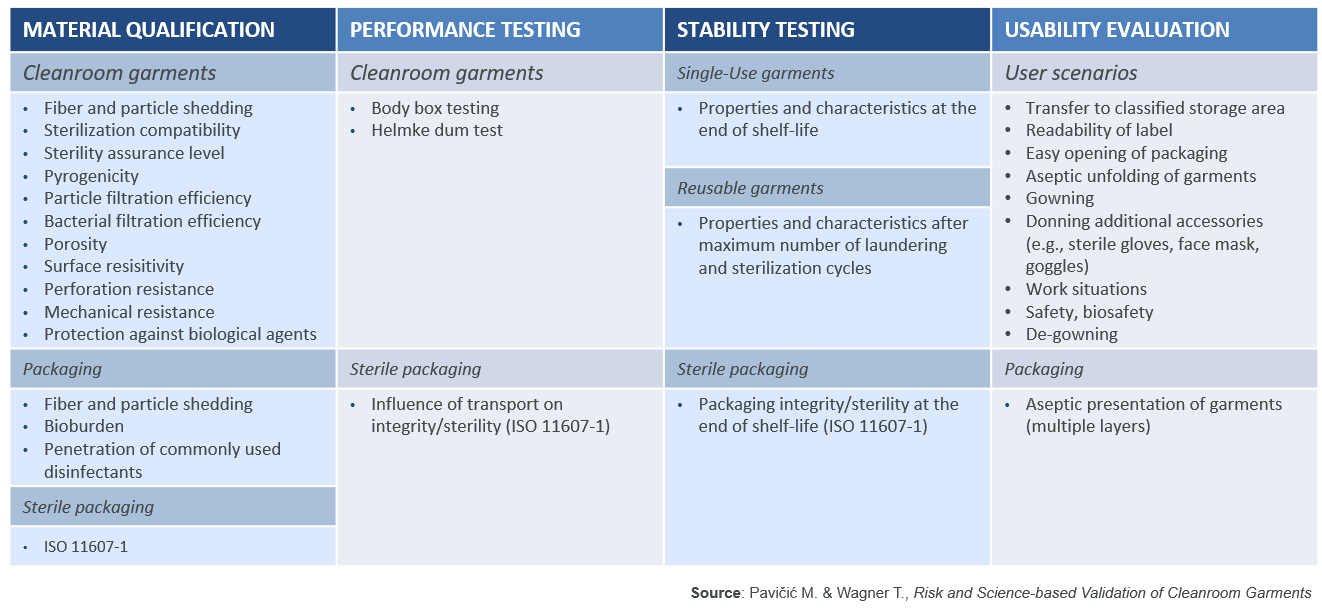
La conformité du système vestimentaire de salle blanche aux cGMP doit être démontrée et documentée lors de la DQ, afin de confirmer que le vêtement de salle blanche sélectionné est qualifié pour l’utilisation prévue. Puisque la nouvelle annexe 1 nécessitera une approche scientifique axée sur les données, la DQ devrait inclure des tests pour simuler l’utilisation prévue et les performances du vêtement. Comme recommandé par la norme ISO 11607-1, la DQ doit être divisée en quatre domaines clés : la qualification des matériaux, les tests de performance, les tests de stabilité et une évaluation des possibilités d’utilisation. Pour les vêtements réutilisables, cela doit être étendu aux sous-traitants, fournisseurs et prestataires de services du fabricant. PAVIČIĆ et WAGNER ont fourni un tableau répertoriant les propriétés qui doivent être évaluées (tableau 1).3
Tableau 1 : validation basée sur les risques et la science des vêtements de salle blanche
Les bioimprimantes basées sur l’extrusion utilisent la pression pour faire passer l’échantillon à travers une buse et créer une forme prédéfinie. Puisque la pression peut facilement être ajustée pour différents types d’échantillons de viscosité, la bioimpression par extrusion est souvent préférée par les chercheurs pour sa flexibilité. Les bioimprimantes basées sur l’utilisation de champs électriques offrent la plus grande précision. Cependant, cela a un prix et les méthodes doivent être rigoureusement optimisées pour éviter de causer des dommages cellulaires.
Dans cet article, seules quelques-unes de ces propriétés seront mises en évidence pour montrer leur importance, ainsi que les méthodes de test scientifiques qui peuvent être utilisées pour évaluer les performances du système de vêtements de salle blanche.
Qualification des matériaux
Afin de vérifier si un vêtement est vraiment stérile, il est important de vérifier si le fabricant suit un processus de stérilisation validé, peut garantir un niveau d’assurance de stérilité (SAL) de 10-6 (selon ANSI/AAMI/ISO 11137-1) et est en mesure de documenter cela dans un certificat de stérilité. Un simple certificat d’irradiation ou un document attestant d’un processus d’autoclavage interne ne suffit pas.
Puisque les vêtements de salle blanche doivent constituer une barrière contre la contamination “humaine” générée par les opérateurs, il est important d’évaluer l’efficacité de filtration des matériaux utilisés dans la fabrication de ces vêtements (par exemple, les tissus en polyester non-tissés ou réutilisables). L’efficacité de filtration des particules (PFE) contre les particules sèches peut être évaluée avec la méthode de test EN 143 (TSI 8130), qui mesure l’efficacité de filtration en utilisant des particules de sel de 0,3 µm de diamètre. L’efficacité de la filtration bactérienne (BFE) peut être évaluée avec la méthode de test ASTM F2101.
Test de performance
Le test du tambour Helmke (IEST-RP-C003.4) est un bon moyen d’évaluer la perte de particules des vêtements de salle blanche, en particulier pour les vêtements lavés plusieurs fois. Pendant ce temps, le test de la “boîte corporelle” (IEST-RP-CC003.4) est la seule méthode de test pour évaluer la perte de particules lorsqu’un vêtement est porté par un opérateur. Nous pouvons ainsi évaluer à la fois le rejet de particules du vêtement et son PFE et BFE par rapport aux particules rejetées par l’opérateur.
Test de stabilité
Il est important de vérifier comment les caractéristiques et les propriétés d’un vêtement changent avec le temps en raison du vieillissement, de l’usure et des cycles de lavage-stérilisation. Par conséquent, les mesures de performances répertoriées ci-dessus doivent être validées dans les pires conditions. Par exemple, pour les articles à usage unique, en évaluant les vêtements de différents lots et en fin de durée de vie ; tandis que, pour les vêtements réutilisables, en évaluant après 10, 20, 30, 40 et 50 cycles de stérilisation lavage-séchage pour déterminer leur performance en fin de vie. Des études menées par ROMANO, LJUNGQVIST et REINMÜLLER ont démontré que des lavages répétés détériorent les performances du vêtement.4,5

Évaluation des possibilités d’utilisation
Il est important de passer par des scénarios d’utilisation et d’évaluer l’emballage des vêtements de salle blanche pour s’assurer qu’ils peuvent être utilisés avec des risques de contamination et de sécurité restants acceptables. Bien que cela soit généralement effectué par l’utilisateur final, les fournisseurs peuvent également effectuer des évaluations et fournir des données aux utilisateurs.
Qualification d’installation (IQ)
Même si l’IQ est un contrôle formel pour vérifier si tous les éléments requis du système vestimentaire de salle blanche sont présents, il est important de vérifier les points suivants afin d’éliminer les risques imprévus :
- Les installations relatives au port et au retrait de la blouse sont-elles en ordre ?
- Le fournisseur a-t-il fourni les certificats de conformité et/ou d’analyse requis, les instructions du fournisseur, etc. ?
- Les procédures opératoires normalisées (SOP) pour le port et le retrait de la blouse ont-elles été rédigées ou adaptées ?
- Les processus logistiques des vêtements et accessoires ont-ils été validés ?
- Les plans de formation et de qualification des opérateurs ont-ils été établis ?
Qualification opérationnelle (OQ)
L’OQ vise à qualifier le concept du port et du retrait de la blouse, y compris la logistique et le passage du matériel, ainsi que la présentation aseptique des vêtements (c’est-à-dire le pliage et l’emballage).
Qualification de performance (PQ)
La PQ est généralement effectuée dans les pires conditions, qui doivent être déterminées sur la base d’une évaluation des risques afin de valider les performances du système vestimentaire pour salle blanche lorsqu’il est utilisé. Les exigences spécifiées dans l’URS doivent être pleinement respectées. Elles comprennent une qualification aseptique de la blouse et une validation de la qualité microbiologique du personnel en blouse, avec vêtements et autres accessoires, lors du travail effectif. Bien sûr, cela ne s’arrête pas là : la revalidation périodique du système de vêtement, la surveillance constante et l’examen critique des modifications apportées au vêtement ou au système sont importants pour démontrer l’état du contrôle.
Conclusion
Les systèmes vestimentaires de salle blanche sont une partie essentielle d’une CCS et de la validation des processus. Une approche basée sur les risques et la science, la qualité dès la conception avec vérification est la bonne stratégie pour contrôler les risques de contamination liés aux personnes et offre des réductions de risques intégrées. Cette approche est une réponse adéquate aux dernières exigences réglementaires.
Références
[1] Ramstorp M (2000) Introduction to contamination control and cleanroom technology. Wiley VCH, Weinheim, Allemagne.
[2] Ramstorp M (2019) Cleanroom garments from a quality risk management approach. Eur J Parenter Pharm 24(3):4–16.
[3] Pavičić M, Wagner T (2019) Risk & science-based validation of cleanroom garments. IVT Network. https://www.ivtnetwork.com/article/risk-science-based-validation-cleanroom-garments
[4] Ljungqvist B, Reinmüller B (2005) Aseptic production, gowning systems and airborne contaminants. Pharm Technol Suppl(2): https://cdn.sanity.io/files/0vv8moc6/pharmtech/75a6ab7e927edd600f5286a1eabb2b41171af658.pdf/article-160408.pdf
[5] Romano F, Ljungqvist B, Reinmuller B et coll. (2016) Performance test of technical cleanroom clothing systems. Proceedings of Indoor Air 2016, 14th International Conference on Indoor Air Quality and Climate, Gand, Belgique.





